Peroxisomal Disorders and Their Treatment
Overview of peroxisomal disorders
Infantile Refsum’s disease (IRD), neonatal adrenoleukodystrophy (NALD) and Zellweger’s syndrome (ZS) are different variants of a group of congenital diseases known as generalized peroxisomal disorders or peroxisome biogenesis disorders (1). These disorders are characterized by the absence of normal peroxisomes in the cells of the body (2). Peroxisomes are small cellular particles that carry out important body reactions, mainly involving fat metabolism. When peroxisomes are lacking, these reactions cannot occur or are defective, and some biochemical abnormalities appear, which give us the diagnosis of the disorder. For example, fatty acids longer than 22 carbon atoms, called very long chain fatty acids (VLCFA), are usually shortened in the peroxisome, and so their levels increase when they cannot be properly degraded due to a peroxisomal defect. Characteristically, these fatty acids are 26 carbon atoms long (26:0 and 26:1), saturated and monounsaturated (without and with a single double bond). Another important abnormality in peroxisomal-disorder patients is a plasmalogen deficiency. Plasmalogens are important phospholipids which are especially concentrated in myelin.
Myelin is the isolating sheath covering the nerve fibers in our brain and nerves; without myelin, conduction of nerve impulses would be very slow. Manufacturing of plasmalogens starts in the peroxisome. So if peroxisomes are defective, plasmalogen levels are low. This can be measured in erythrocytes (red blood cells). In classic ZS, plasmalogen levels are virtually nil. In the milder clinical forms, plasmalogen levels are only marginally decreased. Another diagnostic biochemical sign of peroxisomal disease is the increase in phytanic acid. Phytanic acid derives from phytol, a compound abundant in green leaf vegetables. The synthesis of bile acids is also affected in peroxisomal disorders and some abnormal compounds accumulate.
Peroxisomal disorders are life-threatening diseases, the most rapidly lethal being ZS. These different names do not always coincide with distinct clinical forms, and they vary according to the personal criterion of the physician in charge of the patient. Usually, the less severely affected patients are diagnosed with IRD, but confusion with adult Refsum’s disease, which is a very different disorder, should be avoided. Apart from classic ZS, which is clearly much more severe both clinically and biochemically, generalized peroxisomal disorders form a quite heterogeneous disease group.
Developmental delay and mental retardation is common to all patients, and vision and hearing are affected very soon. Acquisition of speech is particularly affected in peroxisomal patients, even more so than can be expected from their reduced sensorial abilities. On the other hand, in the less severe cases, understanding seems to be relatively preserved in relation to hearing and speech. Due to their reduced communication possibilities, autism is quite common in the patients that live longer.
Peroxisomal-disorder patients practically always present as floppy children, due to their decreased muscle tone (hypotonia), which in the most severe cases is generalized. In IRD, hypotonia is usually restricted to the neck and trunk muscles. Acquisition of head control and independent sitting is often much delayed. Sometimes hypotonia is just revealed by a curved back (kyphosis) in the sitting position. Few patients are capable of walking independently.
Liver is enlarged and liver dysfunction is the rule with corresponding abnormalities in liver enzymes. Very often these patients have fat losses in their stools (steatorrhea). In general, these children are difficult to feed. So failure to thrive is a common characteristic in peroxisomal patients, and the most prevalent presenting symptom in IRD.
Peroxisomal disorders are associated with characteristic facial abnormalities (high forehead, frontal bossing, small face, low set ears, slanted eyes, etc.) However, these characteristics can be very subtle in some children and can be confusing in an infant. So we should not rely on the facial signs alone before suspecting a peroxisomal disorder. Also, while visual and hearing deficits are prevalent signs, they may not appear, or be obvious, in very early infancy. Therefore, for the earliest detection, coincident observations of failure to thrive, hypotonia and psychomotor retardation, widely open fontanel, abnormalities in liver enzymes and enlarged liver should raise serious suspicion of proxisomal disorders.
Treatment of peroxisomal biogenesis disorders
Currently, it is common practice to place peroxisomal-disorder patients on low-fat diets of the type used with adult Refsum’s disease patients (3). This is done with the double aim of lowering the intakes of phytanic acid and VLCFA. Some physicians have also treated peroxisomal patients with a mixture of trioleate-trierucate (4:1 by volume), popularly known as Lorenzo’s Oil, a preparation widely used with X-ALD patients to decrease the levels of VLCFA (4). Some fragmentary experience exists with other therapeutic diets. Plasmalogen precursors (5) and cholic acid (6) have been given to these patients in an effort to increase their plasmalogen levels and normalize bile acids, respectively. A patient treated with plasmapheresis (plasma exchange) was reported to improve in parallel with decreases in plasma phytanate levels (7). Attempts have also been made to improve peroxisomal-disorder patients by providing them with peroxisomal proliferators such as clofibrate, without success (8). No report showing clear evidence of improvement with any of these treatments has been published so far.
Treatment with docosahexaenoic acid (DHA)
DHA accumulation during normal human brain development
Before referring to DHA therapy in peroxisomal patients, it is necessary to mention briefly the developmental changes of this and other lipids in the human brain. During development, brain neurons acquire the rich connectivity that characterizes the human species. This is mainly achieved by increasing the surface of neuronal membranes in the form of specialized receptor arborizations (dendrites) and effector conducting nerve fibers (axons). Axons and dendrites form connections called synapses. Growth of dendrites and synapses is explosive during the last trimester of human gestation, and starts at a definite moment: the 32nd week of gestational age (9, 10). During this time, lipids also increase in a dramatic way, their accretion profiles coinciding exactly with the timing of dendritic growth (11). Fatty acids, which are essential components of lipids, also have developmental profiles that parallel the morphologic changes in the brain. Knowledge of fatty acids is important to physicians and nutritionists because their changes in the diet can modulate the lipid composition of body membranes.
Fatty acids are divided into saturated (without double bonds), monounsaturated (with a single double bond) and polyunsaturated (PUFA, with several double bonds). The importance of the latter comes from the fact that some of them are essential, which means that they cannot be manufactured in the body and must be supplied in the diet. There are two essential fatty acids: linoleic acid (LA, 18:2w6) and a-linolenic acid (ALA, 18:3w3). (After the colon is the number of double bonds, and after the Greek letter w is the position of the first double bond starting from the CH3 group). These two parent fatty acids undergo a series of reactions that produce several long-chain PUFA, the most important of these being arachidonic acid (AA, 20:4w6), derived from LA, and docosahexaenoic acid (DHA, 22:6w3), derived from ALA.
Among long-chain PUFA, DHA is very important because it is particularly localized in brain synapses and in the retina where it can be found at much higher concentrations than in any other place. During development, DHA accretion in the brain is very fast in the third trimester of gestation, and continues at a lower rate until the second year of life (12, 13, 14). In the retina, DHA accretion starts even earlier, at about 24 weeks of gestational age (15). It has been shown that low levels of DHA due to poor w3 fatty acid intakes produce visual and neurological problems in the experimental animal and human subjects (16,17).
Figure 1. DHA accumulation in the normal human brain during development
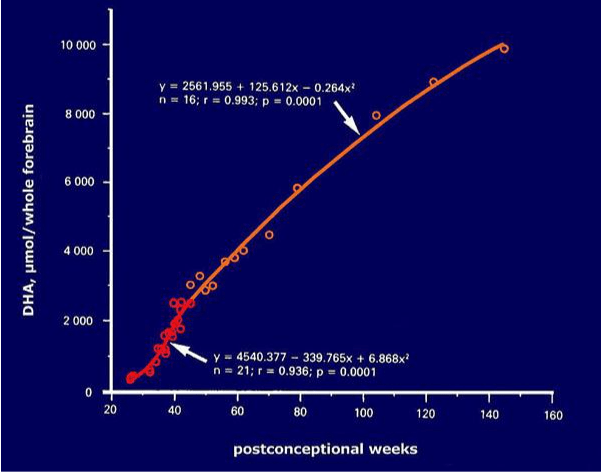
Figure 1 provides a plot of DHA accumulation in the normal human brain during development (13). Ages are given as weeks post conception, and the curve give us the total amount of DHA in the whole cerebrum. Accumulation of DHA is not uniform during this time, so the plot is composed of two curves with different slopes (as indicated by the equations). The red portion of the plot corresponds to the most vulnerable period, that is, the last trimester of gestation and the first weeks of postnatal life. During this time, accumulation of DHA increases rapidly until about one month of life. This is the moment when a poor DHA supply may have the most damaging effects upon the developing nervous system. Later on, from 1 to 18 months of age (orange line), DHA continues to increase at a slower rate. After about two years of age brain DHA practically reaches adult levels.
DHA deficiency in peroxisomal-disorder patients and rationale for the treatment
In 1987, we discovered that the blood, brain, liver and kidneys of a patient with Zellweger syndrome had extremely low levels of DHA (18,19). This finding was later confirmed in other patients with generalized peroxisomal disorders (20, 21, 22, 23, 24). The DHA deficiency was so profound in those patients that it could produce by itself important neurological and visual abnormalities. Indeed, even in the retina, a tissue extremely rich in DHA, the levels of this PUFA were virtually negligible (22).
It is yet unclear why peroxisomal-disorder patients exhibit such a severe DHA deficiency. Are the low levels of DHA the cause or the consequence of peroxisome dysfunction? In this context, it is interesting to note that a link has lately been established between peroxisomes and DHA production in the body. Synthesis of DHA was classically attributed to the endoplasmic reticulum (another important organelle in the cells). Subsequent to the DHA deficiency found in Zellweger’s syndrome, and consistent with that study, a new route for DHA synthesis was proposed by Sprecher’s group (25, 26). According to this route, DHA is manufactured in peroxisomes by b-oxidation of a very long chain PUFA precursor (24:6w3). This new route would provide a mechanism for DHA deficiency in patients with defective peroxisomal b-oxidation and is now widely accepted. Indeed, having a defective b-oxidation of VLCFA, Zellweger patients would be incapable of producing enough DHA. However, patients with other peroxisomal b-oxidation defects do not exhibit any DHA deficiency, and some of the changes found in Zellweger’s syndrome do not totally agree with the new DHA route theory (27).
Whatever the mechanism for DHA production may be, DHA deficiency is always a risk for the development of the brain and retina. In the experimental animal and human, low levels of DHA have been associated with severe visual and neurological damage (28,29). Therefore, we thought that normalizing their DHA levels might be of some benefit to patients with generalized peroxisomal disorders. Thus, in 1991 we started to treat the first two patients with DHA. First of all, we wanted to correct DHA deficiency and normalize things as much as possible. So we chose a pure DHA preparation in order to normalize the DHA levels without altering those of other PUFA. For this reason, mixtures of w3 fatty acids such as those present in fish oil were avoided. They contain high levels of eicosapentaenoic acid (EPA, 20:5w3), an w3 PUFA situated before the probable enzyme block in DHA synthesis. Addition of AA and other w6 PUFA was also avoided since they could compete with DHA and reduce its beneficial effects.
With these ideas in mind, we have treated 25 peroxisomal-disorder patients so far, three of them with classic Zellweger’s syndrome. It is difficult to classify the other 22 patients since there is much confusion nowadays on the subject, as mentioned above. Some of the patients that live longer may appear with time to have different phenotypes to different physicians. This happened with three of our patients. They seemed to have IRD before two years of age, but it was later realized that they should have been diagnosed with NALD. If the patients live long enough, abnormal myelination is finally detected and very often adrenal function is affected. The classic Zellweger phenotype, on the other hand, can be quite clearly distinguished from the others from the beginning. Apart from the extreme severity of their clinical picture, Zellweger patients almost totally lack plasmalogens in their erythrocytes.
Results of DHA therapy
The biochemical results and clinical follow-up of the patients treated with DHA Ethyl Ester (DHA-EE) have appeared in successive publications (30-37). Biochemically, normalization of the DHA levels in plasma and erythrocytes was obtained in all cases. Unexpectedly, plasmalogen levels increased in practically all the patients. In some of them, the 18:0DMA/18:0 ratio (a measure of 18:0 plasmalogen deficiency in these patients) even normalized. It is important to point out that in no case did the plasma VLCFA levels increase due to the normal fat diet provided. On the contrary, both 26:0 and 26:1 decreased in plasma in most DHA-treated, often very markedly so.
Table I. Patients treated with DHA-EE since 1991
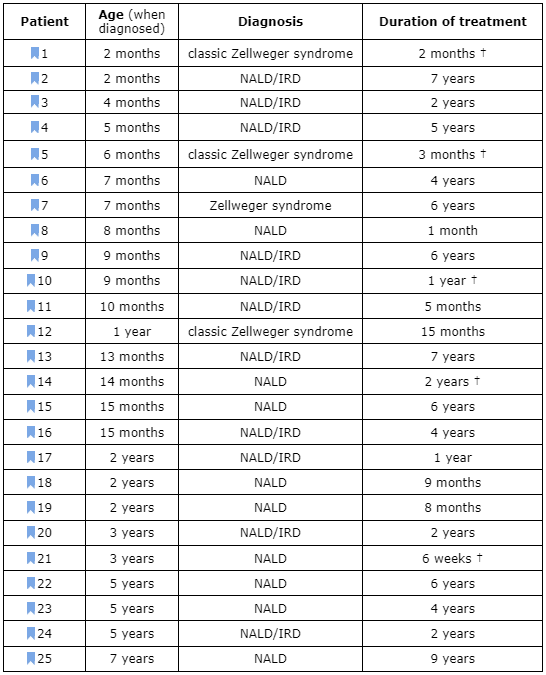
Figure 2 summarizes the most important biochemical and clinical changes in the 25 patients treated with DHA-EE. For reasons mentioned before, the diagnoses are reduced to the “classic” Zellweger’s syndrome phenotype (ZS) and the milder neonatal adrenoleukodystrophy/infantile Refsum’s disease spectrum (NALD/IRD). When one of these phenotypes predominated, it is so indicated in Table I. As can be seen in this table, only 5 patients have died so far. Of these, two had “classic” ZS . A third patient was a 3-year-old girl with a severe NALD phenotype, who was in a terminal, vegetative state when the treatment started and could only be treated for 6 weeks before she died from bronchopneumonia (31). A fourth patient was a girl who had improved quite spectacularly for a year (patient #10) and suddenly died from fulminant septicemia (31,33) unrelated to her disease. Finally, we lost contact with another patient, who suffered from a severe form of NALD and decided to discontinue the treatment (patient #14). The other 22 patients survive. As a whole, this is a much longer survival time than that expected from the spontaneous course of such severe diseases. Besides, most patients have had clinical improvement, at the very least, from the point of view of the nutritional state and liver function. The latter has improved in all patients. Vision has improved in 12 patients, muscle tone in 8 patients and, in general, the patients have become more alert and interactive. Most importantly, myelination has improved in 9 patients, as checked by magnetic resonance imaging (MRI) (34-36). This is the reverse of what normally happens and should be attributed to some still unknown role of DHA in myelin formation.
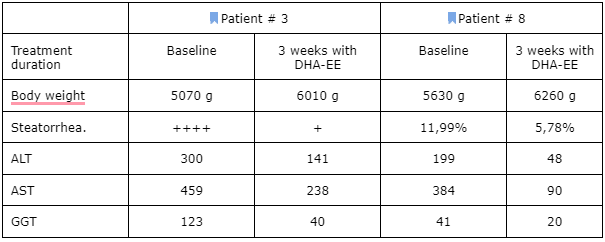
Most significantly, the beneficial effects of the DHA-EE were most marked in three children that started the treatment before 6 months of age. In one of them (patient # 3), it was interesting to find a normalization of intestinal absorption after a short period with DHA-EE. This 5-month-old child had been taking an w3 triglyceride preparation since about a month before the DHA-EE treatment started and an intense steatorrhea (fat loss in the stools) persisted. This totally disappeared after only 3 weeks with DHA-EE. This seems to suggest that DHA-EE may be more effective than other DHA preparations in the treatment of peroxisomal-disorder patients. The same beneficial effect on steatorrhea and liver function has recently been found in another girl with a more severe NALD clinical picture (patient #8). Table II presents these effects in the two patients.
FIgure 2. Summary of the most important clinical and biochemical changes in patients
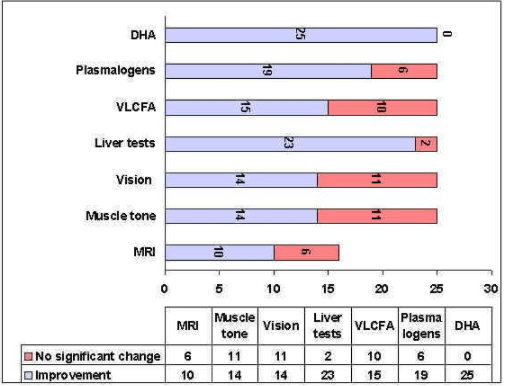
This chart summarizes the most important clinical and biochemical changes in the 25 peroxisomal-disorder patients treated to date. MRI follow-up could only be tracked in 16 of the 25 patients. In some patients treated for a very short period, full benefit of the treatment could not yet be obtained. Being a very rapid response to the DHA-EE treatment, liver improvement is an exception.
Table II: Initial effects of the treatment with DHA ethyl ester in two patients with generalized peroxisomal disorders.
Conclusions
First of all, it must be emphasized that DHA therapy is a nutritional treatment, primarily given to normalize the DHA levels, not to obtain a drug effect. It is important to keep in mind that DHA deficiency always predominates over any other suboptimal PUFA values that may be found. Therefore, DHA deficiency must be corrected first, and for this DHA must be given alone until its blood levels are normalized. This criterion is mainly based on the fact that AA is not decreased in the brain in peroxisomal disease (18–22). Interference with any other PUFA given in high doses, especially with AA, may render DHA therapy ineffective since enzyme competition will favor the w6 over the w3 fatty acid series. Besides, AA is not decreased, but usually increased, in the most important organ: the brain. Therefore, routinely mixing AA to DHA in the treatment of patients with generalized peroxisomal disorders lacks a biochemical basis and may even be dangerous. Indeed, AA will compete with DHA and delay its normalization. In our experience, association of AA to DHA produced an increase in VLCFA and a decrease in the plasmalogen 18:DMA/18:0 ratio in some patients (33). So DHA must be given alone until its levels are normal. Then, if the erythrocyte AA levels fall well bellow their normal range, the diet can be enriched with AA until they are again within the normal. Any change in the diet must always be biochemically monitored.
The brain has a unique opportunity to properly grow and develop and this is during the perinatal and early postnatal periods. Ideally, if we want to correct a DHA deficiency we should provide the DHA during those periods or, at least, as early in life as possible. If we provide the DHA too late, we may correct its deficiency but the past consequences of it may already be irrevocable. Although DHA deficiency is not the only defect in peroxisomal disorders, it is possibly one of the most important abnormalities and, at least, a most serious aggravating factor. So if we want to do the best for these children, their DHA deficiency must be corrected as soon as diagnosed. Besides DHA therapy, peroxisomal patients should receive a nutritious diet and any extra care they need. We must always remember that besides being patients, they are growing children who need everything a child would need for normal development. In our experience, a complete diet does not increase the levels of 26:0 and 26:1 when given with DHA. On the contrary, those levels constantly decrease. So, even if we think that accumulation of VLCFA is the main pathogenic factor in peroxisomal disorders, we should never deprive these children of all the necessary nutrients.
In short, an effort should be made to treat these patients like normal children in every respect, and give them the DHA they need as soon as possible. This treatment may not cure the disease but it will certainly improve the life of these patients a great deal. Logically, when provided very early during brain development, DHA therapy will be the most effective and, perhaps, some brain and visual damage may be prevented. So early diagnosis and early treatment may hopefully change the gloomy prognosis of peroxisomal-disorder patients.
References
- Lazarow PB, Moser HW. Disorders of peroxisome biogenesis. In: The Metabolic Basis of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D (editors). 7th CD-ROM edition, New York: McGraw-Hill, 1997: records 38247-38973.
- Goldfischer S, Moore CL, Johnson AB, Spiro AJ, Valsmis MP, Wisniewski HK, Ritch RH, Norton WT, Rain I, Gartner LM. Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science 1973; 182: 62-64.
- Dry J, Pradalier A, Canny M. Maladie de Refsum: Dix ans de régime pauvre en acide phytanique et phytol. Ann Med Interne (Paris) 1982; 132: 483-487.
- Moser AB, Borel J, Odone A, Naidu S, Cornblath D, Sanders DB, Moser HW.A new dietary therapy for adrenoleukodystrophy: biochemical and preliminary clinical results in 36 patients. Ann Neurol 1987; 21: 240-9.
- Wilson, G.N., Holmes, R.G., Custer, J., Lipkowitz, J.I., Stover, J., Datta, N., and Hajra, A. Zellweger syndrome: diagnostic assays, syndrome delineation, and potential therapy. Am. J. Med. Genet. 1986; 24, 69-82.
- Setchell, K.D.R., Bragetti, P., Zimmer-Nechemias, L., Daugherty, C., Pelli, M.A., Vaccaro, R., Gentili, G., Distrutti, E., Dozzini, G., Morelli, A., and Clerici, C. Oral bile acid treatment and the patient with Zellweger syndrome. Hepatology 1992; 15, 198-207.
- Smeitink, J.M.A., Beemer, F.A., Speel, M., Donckerwolcke, R.A.M.G., Jakobs, C., et al. Bone dysplasia associated with phytanic acid accumulation and deficient plasmalogen synthesis: a peroxisomal entity amenable to plasmapheresis, J. Inher. Metab. Dis. 1992; 15: 377-380.
- Bjorkhem I, Blomstrand S, Glaumann H, Strandvik B. Unsuccessful attempts to induce peroxisomes in two cases of Zellweger disease by treatment with chlofibrate. Pediatr Res 1985; 19: 590-593.
- Martinez M, Ballabriga A. A chemical study on the development of the human forebrain and cerebellum during the brain “growth spurt” period. I. Gangliosides and plasmalogens, Brain Res., 1978; 159: 351-362.
- Martinez M. Myelin lipids in the developing cerebrum, cerebellum, and brain stem of normal and undernourished children, J. Neurochem. 1982; 39: 1684-1692.
- Purpura DP. Morphogenesis of the visual cortex in the preterm infant. In: Growth and Development of the Brain. Brazier MAB (editor). New York: Raven Press; 1975. pp. 33-49.
- Martinez M, Conde C, Ballabriga, A., Some chemical aspects of human brain development. II. Phosphoglyceride fatty acids, Pediat. Res. 1974; 8: 91-102.
- Martinez M. Tissue levels of polyunsaturated fatty acids during early human development, J. Pediat. 1992; 120: S129-138.
- Martinez M., Mougan I. Fatty acid composition of human brain phospholipids during normal development. J. Neurochem. 1998; 71: 2528-2533.
- Martinez, M., Gil-Gibernau, J.J., and Ballabriga, A. Lipids of the developing human retina: I. Total fatty acids, plasmalogens, and fatty acid composition of ethanolamine and choline phosphoglycerides. J. Neurosci. Res. 1988; 20: 484-490.
- Neuringer M, Connor WE, Lin DS, Barstad L, Luck S. Biochemical and functional effects of prenatal and postnatal omega-3 fatty acid deficiency on retina and brain in rhesus monkeys. Proc Natl Acad Sci USA 1986; 83: 4021-4025.
- Holman RT, Johnson SB, Hatch TF. A case of human linolenic acid deficiency involving neurological abnormalities. Am J Clin Nutr 1982; 35:617-623.
- Martínez M. Altered unsaturated fatty acid patterns in Zellweger syndrome. Neurochem Int 1988; suppl 1: 148.
- Martinez M. Polyunsaturated fatty acid changes suggesting a new enzymatic defect in Zellweger syndrome, Lipids 1989; 24: 261-265.
- Martinez M. Severe deficiency of docosahexaenoic acid in peroxisomal disorders: A defect of Delta4-desaturation? Neurology 1990; 40: 1292-1298.
- Martinez M. Severe changes in polyunsaturated fatty acids in the brain, liver, kidney, and retina in patients with peroxisomal disorders, In Neurobiology of Essential Fatty Acids. N. Bazan (editor). New York: Plenum. 1992. pp. 347-359.
- Martinez M. Abnormal profiles of polyunsaturated fatty acids in the brain, liver, kidney and retina of patients with peroxisomal disorders. Brain Res 1992; 583: 171-182.
- Martinez M, Mougan I, Roig M, Ballabriga A., Blood polyunsaturated fatty acids in patients with peroxisomal disorders. A multicenter study, Lipids 1994; 29: 273-280.
- Martinez M, Mougan I. Fatty acid composition of brain glycerophospholipids in peroxisomal disorders. Lipids 1999; 34: 733-40.
- Voss A, Reinhart M, Sankarappa S, Sprecher H. The metabolism of 7,10,13,16, 19-docosapentaenoic acid to 4,7,10,13,16,19-docosahexaenoic acid in rat liver is independent of a 4-desaturase. J Biol Chem 1991; 266: 19995-20000.
- Moore SA, Hurt E, Yoder E, Sprecher H, Spector AA. Docosahexaenoic acid synthesis in human skin fibroblasts involves peroxisomal retroconversion of tetracosahexaenoic acid. J Lipid Res 1995; 36: 2433-2443.
- Infante JP, Huszagh VA. On the molecular etiology of decreased arachidonic (20:4n-6), docosapentaenoic (22:5n-6) and docosahexaenoic (22:6n-3) acids in Zellweger syndrome and other peroxisomal disorders. Mol Cell Biochem 1997; 168: 101-115.
- Neuringer M, Connor WE, Lin DS, Barstad L, Luck S. Biochemical and functional effects of prenatal and postnatal omega-3 fatty acid deficiency on retina and brain in rhesus monkeys. Proc Natl Acad Sci USA 1986; 83: 4021-4025.
- Holman RT, Johnson SB, Hatch TF. A case of human linolenic acid deficiency involving neurological abnormalities. Am J Clin Nutr 1982; 35:617-623.
- Martinez, M. Treatment with docosahexaenoic acid favorably modifies the fatty acid composition of erythrocytes in peroxisomal patients. In PM Coates and K Tanaka (Eds) New Developments in Fatty Acid Oxidation, Wiley-Liss, New York, 1992; 389-397.30 Martinez, M. Treatment with docosahexaenoic acid favorably modifies the fatty acid composition of erythrocytes in peroxisomal patients. In PM Coates and K Tanaka (Eds) New Developments in Fatty Acid Oxidation, Wiley-Liss, New York, 1992; 389-397.
- Martinez, M.., Pineda, M., Vidal, R., and Martin, B., Docosahexaenoic acid – A new therapeutic possibility to peroxisomal-disorder patients: Experience with two cases, Neurology 1993; 43: 1389-1397.
- Martinez M. Polyunsaturated fatty acids in the developing human brain, erythrocytes and plasma in peroxisomal disease: therapeutic implications. J Inher Metab Dis 1995; 18 Suppl. 1, 61-75.
- Martínez M. Docosahexaenoic acid therapy in DHA-deficient patients with disorders of peroxisomal biogenesis, Lipids 1996; 31: S145-152.
- Martinez M, Vazquez E. MRI evidence that docosahexaenoic acid ethyl ester improves myelination in generalized peroxisomal disorders. Neurology 1998; 51: 26-32.
- Martinez M, Vazquez E, García Silva M.T, Manzanares J, Bertran JM, Castelló F, Pineda M, Mougan I. Tratamiento de las enfermedades peroxisomales generalizadas con etil ester del ácido docosahexaenoico. Rev Neurología 1998; 28: S59-64.
- Martinez M, Vazquez E, García Silva MT, Manzanares J, Bertran JM, Castelló F, Mougan I. Therapeutic effects of docosahexaenoic acid in patients with generalized peroxisomal disorders. Am J Clin Nutr 2000;71: 376S-85S.
- Martinez M, The fundamental and practice of docosahexaenoic acid therapy in peroxisomal disorders. Curr Opin Clin Nutr Metab Care 2000; 3: 101-108.
Biography of Dr. Manuela
Manuela Martinez was a doctor of medicine, a pediatrician and a biochemist working in the field of lipids and human brain development since 1972. She was been mainly interested in the effects of nutrition with different diets and proportions of fatty acids on early human development. She started by studying the fatty acid composition of phosphoglycerides in the developing human brain during the prenatal and neonatal periods. She then studied the lipid changes during the period of rapid myelination in whole brain tissue and also in the pure myelin fraction. In contrast to the rat, the galactolipid profiles showed that the forebrain myelinates faster and is more vulnerable to malnutrition than the cerebellum. Another unexpected finding was that myelination in the human brain proceeds by the accretion of myelin, biochemically quite mature from the beginning. As a consequence, nutritional aggressions affect the amount rather than the quality of myelin, a fact that was in contradiction with what was expected from studies in the experimental animal. The lipid and fatty acid accretion in the developing human brain starts to accelerate markedly after 30-32 weeks of gestation. These changes correlate with the fast formation of synapses and dendritic spines found by other authors. In the human brain, the accretion of polyunsaturated fatty acids (PUFA), especially docosahexaenoic acid (DHA, 22:6w3), which has a parabolic profile and is maximal between 30 weeks and the end of gestation, but continues during postnatal life until about two years of age. More easily than in the rat, in the human species malnutrition and administration of diets with unbalanced omega-3 to omega-6 ratios may lead to suboptimal levels of DHA in the retina and other tissues.
Her previous work led Dr. Martinez to discover a new abnormality in patients with Zellweger’s syndrome, a congenital disease characterized by the lack of functioning cell peroxisomes. The new abnormality found was a deficiency of the most important omega-3 polyunsaturated fatty acid in the brain and retina: docosahexaenoic acid (DHA, 22:6w3). Since some of the signs and symptoms that these patients exhibit are similar to those observed in the experimental animal model deprived of omega-3 fatty acids, the DHA deficiency found might be related to the cause of Zellweger’s syndrome. With this rationale in mind, in 1990 Manuela Martinez devised a new treatment for these patients, based in the correction of their DHA deficiency. This treatment is producing significant beneficial effects in patients with relatively mild variants of Zellweger’s syndrome, and is currently been used by several other physicians in the world. For this work on DHA and Zellweger’s syndrome, Dr. Martinez was awarded Spain’s Queen Sofia’s Prize for Research on the Prevention of Deficiencies in 1998.
Contact Information
Dr. Manuela Martinez, M.D.
Stauros Children’s Clinic
Plaza Karl Marx, s/n
08035 Barcelona, Spain
Teléfono: (+34) 93 427 6000
e-mail addresses: 3572mmr@comb.es
—————————————–
Manuela Martinez Foundation
Gran Vía, 750, 6-1
08013 Barcelona, Spain
Teléfono: (+34) 93 231 6238